In this section, you will learn how to gather a group of related sequences in FASTA format, and then use them as inputs to the program ClustalW. The result is a multiple-sequence alignment (MSA), from which you can deduce much about how the sequences resemble and differ from each other. Then you will use the MSA as input to tree-printing programs, in order to produce a phylogenetic tree—a visual summary of relationships among the genes.
Answering this question requires making a multiple sequence alignment and then using it to make a phylogenetic tree. For these tasks, you move to another database where it's a little easier to gather a bunch of sequences into a single FASTA file.
Point your browser to http://www.expasy.ch/.
You see the home page of ExPASy, the Expert Protein Analysis System. As stated in the Cast of Characters, ExPASy is a complete protein tool box. With ExPASy, you can do almost any imaginable analysis or comparison of protein sequences and structures. In my humble opinion, Swiss sequence database tools are among the easiest ones to use.
Click UniProt Knowledgebase (SwissProt and TrEMBL) under Databases.
Read the introduction to these databases. They are high quality protein (not nucleic acid) sequence databases with abundant annotation, minimal redundancy, and many connections to other databases.
Click New UniProt Website. The new (2008) home page of UniProt contains links to information about the resource. Click to learn more about the site, and then return to this page. Bookmark this page (UniProt Welcome) as a good starting point for future use of UniProt, Swiss-Prot, or TrEMBL.
At the top of the page is a deceptively simple but powerful search tool. A menu lets you choose among data sets to search. Take a look at the list on the menu, put return it to Protein Knowledgebase (UniProtKB).
In the Query box, type opsin. Click Search. The search produces over 4000 entries, all of which are protein entries that are opsins or include the word or fragment -opsin-. Obviously, you need to be more specific.
Limit the search to human opsins, as follows. Click Fields, beside the Query box. The Search area expands to include a logical operator menu (with default operator AND), a Field menu, and a Term box. Under Field, pick Organism. In the Term box, start typing human. As you type, the search tool helpfully shows you all allowed search terms that fit what you have typed so far. As soon as human [9606] appears, click it to enter it in the Term box, and click Add and Search.
Notice that the Query box now says "opsin AND organism: human [9606]". This shows that you have limited your search to opsin-related entries that are also (AND) human proteins. Notice also that the Fields link is available again, so that you could add additional terms to your search, with logical operators AND, OR, and NOT to specify how to use the additional terms. But the search is already specific enough to make our task easy: there are only 25 results for this search.
Before looking at the results, look at the other Fields you can search. UniProt entries are files that are divided into sections, called fields, each containing specific kinds of information. You can limit searches to terms that reside in specific fields, or can simply search for your query in entire entries.
Now look over the results. On 2008/09/19, this search gave 25 hits, including the rod pigment rhodopsin (OPSD), along with the three cone pigments (OPSB, OPSG, OPSR). There is also a "visual pigment-like receptor peropsin", OPSX, which still, more than ten years after its discovery in the genome, is of unknown function. In the rest of this tutorial, you will include this mysterious protein in your inquiries into the visual pigments of the human retina.
Now you will digress briefly from the question of how these proteins are related evolutionarily, and find out more about peropsin. In the process, you will glimpse the wealth of information in, and linked to, a typical UniProt entry.
In the Accession column, click O14718, next to OPSX_HUMAN.
By the way, an accession number such as O14718 can be used as an iput to almost any ExPASy tool for analysis of the corresponding sequence.
You see the UniProtKB View of entry O14718 [note: that first character is capital letter O, not zero (0)]. Peruse this entry and try to find out just what this rhodopsin-like protein is thought to do. Under General annotation (Comments), you'll learn that it is found in the retina (the RPE or retinal pigment epithelium), and that it may detect light, or perhaps monitors levels of retinoids, the general class of compounds that are the actual light absorbers in opsins. Also under Similarity in the same section, you see, as mentioned earlier, that this protein is a member of the large family of G protein-coupled receptors (GPCRs). If you click G-protein coupled receptor 1 family, you conduct a search for a members of this family—the result is about 10,000 hits in UniProt. Limit this search to humans (about 1200 hits). Back on the O14718 page, click Opsin subfamily to find a list of all purported members of this subfamily in UniProt (about 220). Limit the search to humans (fewer than 20).
Once again, back up to the UniProtKB entry page for O14718.
Under References find the journal citation, "Peropsin, a novel visual pigment-like protein located in the apical microvilli of the retinal pigment epithelium.". Click the PubMed link with that reference to see an abstract of the paper. On the abstract page, click on of the Free Full Text Article links to obtain the full paper from either the journal (PNAS) or from PubMed Central, which distributes many articles. Like many journals, PNAS puts full articles online just 6 to 12 months after publication.
Return to O14718, and look around more on the entry page. You will find Cross-references to this protein or its gene in other databases, predicted structural features of the protein, and the sequence, which you can lift in FASTA format if you wanted to search for more of its relatives. Note also links to a number of ExPASy tools listed for further analysis of this sequence.
Try one of them: under Cross-references, find PROSITE, and click Graphical view.
You now have a form that allows you to search for signatures of function or functional sites in peropsin. Leave all settings as they are, and click scan next to the graphical image (green) of the protein. Here is another form, with the accession number O14718 already entered. Again, leave all other settings as they are (but notice that there are many ways to modify this search), and click START THE SCAN.
PROSITE finds three identifiable things about this sequence. One "hit by profile" identifies peropsin as a G-protein coupled receptor. Two "hits by pattern" are shown. One is a short sequence that also identifies peropsin as a GPCR, while the second hit identifies a binding site for retinal. So PROSITE indicates that, like its visual opsin relatives, peropsin also binds specifically to retinal, the visual pigment that we make from vitamin A. Note also that, by similarity to other related proteins, PROSITE predicts the presence of a disulfide bond, between residues 98 and 175.
(Later, you will find out more about the three-dimensional structure of peropsin by building a model of it. You will use a related protein of know structure as a template for making this model. This process is called homology modeling.)
Next you will answer the main question of this section: how are the visual pigments (and peropsin) related to each other? Apparently, they diverged from a common ancestral opsin, but you can get a much clearer picture of which of these opsins came first, and which are the most closely related. To answer this question, you will align all their sequences (called a multiple sequence alignment) and then produce a little family tree. UniProt provides easy access to ClustalW, which does multiple-sequence alignments in a snap, as well as the information needed to print a phylogenetic tree from the alignment information.
Return to the UniProt search results, with its 25 hits for entries from the human genome that include the description "opsins". Your next task is to compare the sequences of peropsin and four visual pigments. Start by clicking to put check marks in the left-hand column of the results table, beside the first four entries (rhodopsin and the blue-, red-, and green-sensitive opsins) and also in the row for peropsin, O14718. As you put in the first check mark, a green band appears at the bottom of the window, providing a tool bar with options for handling multiple sequences. After you have checked the entries as instructed, click the Align button in the green tool bar. This is a request to use ClustalW to make a multiple-sequence alignment using the selected entries.
The Clustalw results page appears. At the top, in the Sequences box, are FASTA-format listings of all the sequences compared. Take a moment to edit this listing to make subsequent alignments and trees easier to interpret. In the FASTA sequences listed in the Sequences box, make the follow changes:
After editing, click Align to redo the alignment with new headings.
To save this alignment in a form needed for the next section, click the orange TEXT button to the right of Clustalw Results. Copy the text file that is displayed, paste it into a new text file, and name it OpsinMSAEdited.txt. Now back up to the Clustalw Results page.
Below the table that names each opsin with your new headings is the multiple sequence alignment. In blocks of 60 residues, Clustalw has aligned five sequences. Below each column of five residues, symbols indicate how closely the residues match across the five proteins. "*" means all 5 aligned proteins have the same amino-acid residue in this position (fully conserved residues, within this group); ":" means that all residues in this position are very similar in size, charge, and polarity (replacements are very conservative); "." means that they are sort of similar (somewhat conservative replacements); and no symbol means that the residues in that position vary greatly in properties (nonconserved residues). (What does each symbol suggest about the importance of that residue to the function of this protein family?)
At the bottom of the results page are several tool bars. Play with the first two to see what they do. You will find that they modify the display of the multiple-sequence alignment to highlight residues types or signatures of protein function. Using these tools, you can get a general picture of similarities and differences among the proteins. But the comparison can be made much more explicit by using it to make a phylogenetic tree for this group of proteins. The last tool bar provides a ClustalW tree. You will learn more about the meaning of various types of trees below.
As you can see at the bottom, this page also provides the information needed to print a tree with more flexibility, and a tool at the University of Indiana can use that information. Unfortunately, this tree is not a true phylogenetic tree; it is a simple tree that shows the order in which ClustalW carried out pairwise alignments as it built the multiple-sequence alignment. It will show the pairs that are most closely related to each other, but you must use a more powerful tree-generating program to obtain a more rigorous tree.
NOTE: This type of ClustalW working tree file always has a .dnd suffix. For really good phylogenetic trees, do not use .dnd files.
Anyway, we can use this tree just to learn how to print trees once you have a good one from any source (next section). This procedure will work if you have tree data in Newick format, which is true for the tree file provided on this page. Get the file you need to make a tree by going to the top of the page and clicking the orange TREE button. Your browser display a very small text file, littered with parentheses. Copy and save this file as ClustalwTreeData.txt. This is tree data in Newick format, a widely used format for tree-printing programs. You will use the data in this file to print your first tree.
A convenient tree printer, Phylodendron, is located at http://iubio.bio.indiana.edu/treeapp/treeprint-form.html. When you point your browser to this URL, you find the input form for the phylogenetic tree printer.
Paste the contents of your ClustalwTreeData.txt into the Tree data box near the top of the form. Type a title into the Title box, something like "Opsin Family Tree". To get a tree that looks like mine (below), pick Phenogram from the Tree styles at the top. Then under Extra Options, select Format: GIF image; width and height: 400 pixelsFont: Helvetica; Style: plain; Size: 12. Leave all other settings as you found them, and click Submit.
Your tree should appear in your browser. Save it OpsinTree.gif. Be sure to remove ".cgi" from the default name, so that your file will be recognizable as a normal GIF file. You can paste these files into documents for reports and publications. Play around with other options at Phylodendron, and see how they affect the tree image.
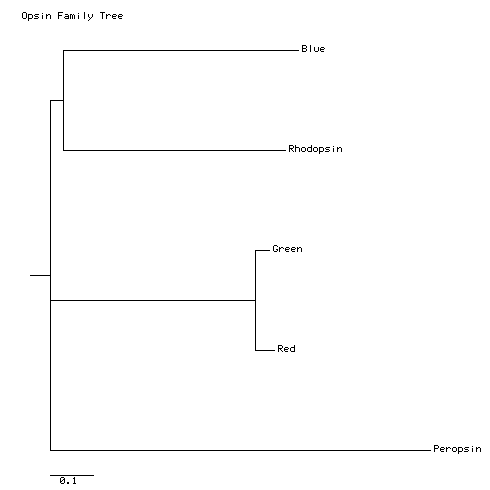
With the settings given above, my tree looks like this:
In a true phylogenetic tree (this is not), the horizontal dimension is time. The vertical dimension is extent of sequence change. Each tip represents a sequence at the present time. Each fork represents an ancestral sequence, and an event of divergence between two current sequences. The horizontal distance between a fork and the tips of the fork represents the time since divergence, and the vertical distance between tips represents the amount of sequence difference between the tips.
Like this tree, most trees produced by bioinformatics tools are unrooted trees; that is, the tree shows distances, based on sequence differences, between the tips, but it does not attempt to show the tips and branches in order of their appearance in time. Sequence-comparison programs cannot figure out the order or direction of evolution. They can only assess the magnitude of sequence differences. If you know which sequence is the progenitor of all the others (we don't, in this case), you can root the tree with that sequence. The result will be that the first branch will separate that sequence from the others. The tree above happens to be rooted with peropsin, so it shows the first branch as the divergence of peropsin from the progenitor of all the other opsins. More advanced tree-building programs allow you to choose the root sequence for a tree, but remember that sequence information alone will not tell you the root.
The conclusions of the previous paragraph are based on examining this printed tree. We will see later that this tree is very similar to a tree made by a more rigorous method. This simply means that this particular tree is an easy one to determine. Most trees are not so easy, and more rigorous methods will give results that are substantially different from ClustalW's little working .dnd file.
Remember also that the truth of any conclusions drawn from a tree depends on the accuracy of the multiple sequence alignment and on the alignment scores. In this tutorial, you are using default settings on many hidden parameters in the processes of comparing and aligning sequences. If you want to draw conclusions about phylogenetic relationships that will hold up to scientific scrutiny, you need to learn much more about the inner workings of alignment tools like Clustalw.
In the next section, you will make this tree two more times, using more rigorous tools for calculating phylogenetic distances.