9. Judging the Quality of Models
The Protein Data Bank is the world's main
repository for experimental models, which means models derived
from one of two methods: analysis of x-ray diffraction by crystals (a
method called x-ray crystallography), or nuclear magnetic resonance
(NMR) spectroscopy. A few other models currently residing at PDB are
theoretical models, such as homology models, which are built
by threading a sequence onto an x-ray or NMR model of one or more
homologous proteins. Automated projects have produced massive
databanks of homology models, like SWISS-MODEL Repository.
Theoretical models also result from other areas of structural
research, such as attempts to predict conformation from sequence, or
to simulate protein dynamics.
Your most important quality-control action is to realize that all "structures"—whether from X-ray crystallography, NMR, homology modeling, or other forms of modeling— are models; they are not observed molecules. Models might harbor many kinds of errors, from minor and inconsequential conformational errors to massive blunders (including complete misalignment of the sequence with the the X-ray electron density or the NMR constraints]). Finding errors requires comparing structural properties of a model with expected structural properties of molecules, as well as making full use of all the information (not just the coordinate list) included in a model file from the Protein Data Bank.
All methods of obtaining models of macromolecules
produce more than merely the lists of coordinates that DeepView uses
to produce graphics models. Among other information, experimental
studies yield evidence about the amount of disorder or mobility in
various regions of the molecule, and they yield statistics about the
precision of the atom positions in the model. Homology modeling
results include valuable information about how well each region of
the model fits its templates, and thus how much confidence is
justified. If you plan to use any macromolecular model to help you
interpret the results of chemical, kinetic, thermodynamic, or other
kinds of studies on a molecule of interest to you, you need to use
wisely all the information that accompanies a new molecular
structure, and you need to be sure that you are working with a
high-quality model.
In the three parts of this section, you will learn
to use some features of DeepView that help with this task,
including, for crystallographic models, coloring the model by
B-factors and examining the quality of the electron-density map that
was used to produce your model; for NMR models, examining the
extent to which multiple models agree with each other; and for homology models, making them and then assessing whether they fit with general expectations about protein structures, such as whether specific residues are in expected environments (for example, in a water-soluble protein, non-polars buried, polars exposed).
NOTE: Many of the tools used to judge quality of homology models will also find problems in X-ray and NMR models. So I have introduced tools that are useful for all types of models in the last section, on homology models. Even if you do not plan to make or use homology models, you should work through section 9c to learn about the full range of model-quality tools in DeepView.
9a. Judging the Quality of Crystallographic
Models
First, read this Technical
Note: How Structures Are Determined From Diffracted
X-Rays.
For this section, you will need two new files. The
first, 1HEL.pdb contains the coordinates of lysozyme without tri-NAG.
The second, 1HEL.dn6 is an electron-density map, the molecular image
obtained from x-ray crystallography. To obtain these files, click the
appropriate one of these links:
You will receive a folder named HEL. Inside the
folder, you will find the files described above.
Start DeepView, but click Cancel on the
dialog box for loading files. Even though you did not load a file,
you still see the DeepView menus at the top of the
screen.
Prefs: Loading Protein...
On the resulting dialog box, uncheck the last box,
Ignore Solvent.... This action tells DeepView not to ignore,
but to include, solvent (usually water) molecules when it
loads a file. Not all PDB files contain solvent molecules, but as
described in the technical
note, the better models do. Make sure that
the following options are checked: Show Solvent and
Show Hydrogens. Make sure Center It is not
checked.
NOTE: Prefs: Loading Protein...
gives you many ways to modify the initial display of protein
models. If you carry out the same display operations almost everyt
time you load a model, you may want to change some of these
preferences so that DeepView can do the work for you.
Now load the pdb file 1HEL.pdb. Select, display,
and center the full model.
Notice the red crosses all around the protein.
Each cross shows the location of an ordered water molecule. Water
molecules are located during structure determination by x-ray
crystallography.
Wind: Layer Infos
Click to remove the checkmark under HOH. Click again to replace
it. This is a quick way to turn on and off the display of solvent
molecules. For now, leave the water molecules off. Notice that the
checkmark in the HOH column controls display (show) of HOH,
but does not control selection.
Color: B Factor
You should see the model in colors ranging from blue
to orange, as you did when you colored by accessibility. This time,
colors are based on numbers called temperature factors or B factors
found in the PDB file. These numbers result from crystallographic
structure determination. They tell, for each atom in the model, how
well determined is its position. Positions of atoms colored dark blue
are the most certain, while those in red are least certain. Atom
positions can be uncertain because of disorder in the crystal from
which the structure was determined. In a high-quality model, the B
factors reflect the mobility or flexibility of various parts of the
molecule. Red residues are the most mobile or "hot," while blue are
the most immobile or "cold."
If you color backbone+side by B factor, mainchain
color is determined by the highest B factor of mainchain N, CA, or C,
while sidechain color is determined by the highest B factor in each
sidechain. As you can see, B factors are lower for main-chain than
for side chains, especially those on the surface. Identify some of
the side chains with high B-factors. Are most of these "hot" residues
buried or on the surface? Use a slab with a depth of 8 angstroms to
help you answer this question.
Use the Tool Bar's label button (marked
"LEU 41?") to identify and label a residue with high B-factors.
Remember the residue number. Press escape to turn off
labeling. Now click the file button (the dog-eared page symbol
below the screen attributes button -- first button in upper left) to
view the PDB file. Scroll to the residue number you selected (residue
numbers are in the fifth column, following the residue name). Look at
the B-factors for atoms in the residue -- they are in the last column
before "1HEL". You will most likely see lower B-factors for mainchain
atoms than for sidechain atoms. Compare the B-factors for atoms in
the side chain you chose with those of nearby residues. Notice the
full range of values. The lowest values correspond to the least
mobile or best determined parts of the molecule. Values above 60 may
signify disorder (or in rare cases, errors in the model), or in NMR
models, large discrepancies among the various models that fit the NMR
data.
Close the file window. Remove any labels by
choosing Display: Labels: Clear User Labels. Turn off slabbing
if you have been using it. Color the full model in CPK
colors.
File: Open Electron Density Map: DN6
In the dialog, find the file 1HEL.dn6, select it, and click
Open. A large dialog appears. Click to darken the button
labeled Display Around CA. You should find the number 7.5 in
all three boxes adjacent to this button. If not, use tab to place the
cursor in these boxes and replace the value shown with 7.5. You are
limiting the display of the electron density map to within 7.5
angstroms of the alpha carbon atom (listed as CA in PDB files) in the
currently centered group. Then click OK. There will be a delay
while DeepView loads this large file. When the graphics window is
active, press help or = to center the view. You will see a
cloudy, deep red image on part of the model. This is the
electron-density map (EDM), essentially an image of the electron
clouds surrounding the atoms of this protein.
Wind: Electron Density
This window allows you to control the map display. Place this
window in a convenient location.
In the Control Panel, find PHE38.
Option-click (right mouse button on Windows) on its name. This
action centers CA of this residue in the display, and it also
calculates and displays a section of the map centered on this CA
atom. Zoom in to get a close view of the map and model
(stereo
recommended). The map is the image of the molecule that was obtained
from x-ray crystallography. If you cannot see the map well, change
its color in Prefs: Electron Density Maps, the same dialog
that appeared when you loaded the map. Click the top Color
button to select a new color -- try brightening the red
slightly.
NOTE: DeepView turns the map display off
when you move the model. This simplification of the display makes
for quicker, smoother movement. If you have a very fast computer,
you can keep the map visible as you rotate. Use Prefs: Real
Time Display to determine what features of the display are off
or on during movement. You may also need to increase the number of
lines displayed before simplification occurs. (On a fast computer,
set it for 1,000,000 lines.) How do you know if you have a fast
computer? Set the preferences to keep the map on display during
movement. If movement of the map is smooth, you have a fast
computer. If it's very slow and choppy, you don't.
The model you are viewing was actually built to
conform to this map. Because electrons, but not atomic nuclei,
diffract x-rays, the x-ray image is that of the electron clouds
around the atoms. The map is drawn (or "contoured") to define the
surface at which the electron density has a constant value, the same
way that atomic orbital diagrams in chemistry textbooks are drawn to
show the volume within which the probability of finding the electron
is constant, say, 90%.
Use an 8-angstrom slab (Display: Slab --
and you can change slab depth with Prefs: Display) to simplify
the view, and zoom in close. Rotate the model to see the PHE ring
edgewise. This will show you how the model conforms to the map. In
this region, the map is well-defined. Select the entire model and
color it by B-factor. The model colors in this region are blue,
signifying small B-factors.
Press the cursor-left key (labeled
<--). This action moves the center of the display, including the
map, down one residue to ASN37. (You can use
cursor-left and cursor-right to move
through the model one residue at a time, as crystallographers would
if they were systematically checking and rebuilding the model to fit
the map better.) The side chain of ASN37 has a higher B-factor
(colored yellow-green). Notice that part of the model extends out of
the map. In other words, the map does not clearly define the
positions of atoms with higher B-factors.
Remember that a crystallographic model shows the
average molecule in a crystal. If, for example, an ASN side
chain is in one conformation in some of the lysozyme molecules in the
crystal, and in another conformation in other molecules, the observed
ASN electron density will be weakened. When there are two
well-defined conformations, the map may show both conformations as
low density. Some PDB files contain coordinates for two alternative
conformations of such residues.
Press the cursor-down key once and
watch the map, especially at the tip of the ASN37 side chain, where
the model extends beyond the map. Press again and again until the map
encloses the tip of the chain. With each press of the key, you are
contouring the map at a lower value of electron density; that
is, you are moving the map surface to show lower values of electron
density. Where the molecule is disordered, the electron density
appears to be lower. The current coutour level is shown in the
Electron Density window.
The active-site residues of lysozyme are GLU35 and
ASP52. Are these important residues well-defined by this map? This
would be an important question if you were planning to base an
interpretion of lysozyme's action upon the precise positions of atoms
in this region of the model.
Center the display on GLU35. Notice small balls of
electron density floating in the vicinity of the GLU side chain. Use
the Layer Infos window to turn on display of water molecules (HOH
column). Each red cross appears at the proposed location of the
oxygen atom of a water molecule. The presence of water is inferred
from the balls of density. Their appearance in the electron-density
map inplies that these waters must be immobilized on the protein
surface in most of the molecules in the crystal.
Now compute H-bonds and use the Layer Infos window
to turn on the display of hydrogen-bond distances (Hdst column). You
can see that the water molecules in this area are within reasonable
hydrogen-bonding distances of atoms on the protein surface, or of
other waters. If you see any waters that are not surrounded by
electron density, lower the contour level of the map by pressing the
cursor-down key. Keep pressing it until electron density appear. A
water molecule that exhibits weak density may be present at the
indicated location in only a small percentage of the lysozyme
molecules in the crystal.
Lysozyme hydrolyzes its substrate (cleaves it with
water). GLU35 is proposed to participate in this cleavage. Perhaps
one of the waters in this region of the model occupies the site of
the water molecule involved in catalysis.
Finally, examine the PDB file for 1HEL by clicking
on the document icon just above the graphics display. In the header
(comments before the ATOM lines), find the R-factor for this model,
and the average deviation (RMSD) of bond lengths and angles from
expected values. The technical
note for this section tells more about
these indicators of model quality.
Also notice another handy feature: in the PDB file
display, click on CA of any residue. The entire atom line turns red,
indicating that it is selected. Then close the window. You will find
that DeepView has centered the display on the CA you chose,
contoured the map with the same center, and limited the display to
atoms within 7.5 anstroms of the center. You can change this distance
limit, called the Auto Center & Inspect Radius, with
Prefs: Display.
In one of the ADVANCED TUTORIALS (see Contents frame: Fitting Residues into Electron Density) you can explore EDM's further, by
trying to identify sidechains from the map alone, and then building
side chains to fit the map.
9b. Judging the Quality of NMR Models
First, read this Technical
Note: How Structures Are Determined By NMR
Spectroscopy.
Obtain the files,
1BCN
and 1BBN
directly from the Protein Data Bank. 1BCN contains an ensemble of 22
NMR models of interleukin 4, a small protein involved in regulating
immune and inflammatory responses. All 22 models fit a set of
NMR-derived constraints that were used in structure determination.
1BCN is a large file, and may take a few minutes to download. File
1BBN contains one model of interleukin 4, derived by averaging the 22
models in the ensemble file, and then energy-minimizing the
result (called an averaged, energy-minimized model).
NOTE added 2008/07/02: Most NMR groups no longer deposit averaged, energy-minimized models, like 1BBN. They recommend that users study the entire ensemble to get the most complete picture of what the NMR modeling reveals. In averaged models, you can easily find atoms in positions that do not agree with any of the models in the ensemble. So averaging introduces features that are not supported by experimental evidence.
File: Open PDB File...
Find and open 1BCN. DeepView informs you that the file contains
more than one model, and asks how many to display. The default number provided is the actual number in the file. Open all 22 models.
The resulting display resembles a porcupine. You are looking at 22
similar models, all superimposed on each other. DeepView loads the
models into separate layers. Remember that all of the models in an NMR ensemble fit the data. So we cannot say that one is correct; nor can we say that an average of all models is the correct model. The variation in the ensemble reflects uncertainty in the structure determination, just as B-factors reflect uncertainty in crystallographic models.
Uncertainty is a part of all molecular models. (Sign at a nearby doughnut shop: "Life is uncertain. Eat dessert first.")
Use the Layers Info window to turn off H in all layers (shift-click a check mark
in the H column), and observe the result. Unlike most
crystallographic models, NMR models contain hydrogen atoms. While the
resolution of most—but not all!—protein electron-density maps is
too low to reveal hydrogen atoms, NMR models are built largely on the basis of measured distances and dihedral angles between hydrogen atoms.
Use Layers Info to display all models as
alpha-carbon traces (CA column). Then use the Control Panel to hide
all side chains, as follows: hold down shift and ctrl, and click any checkmark in the side column. Then blink continuously (hold down shift and tab) to see the amount of variation in the models in this large ensemble.
Again, all of these models fit the NMR data. In regions where the models agree with each other, we might say that the structure is well-determined, or that there is low uncertainty in atom positions. In regions where the models exhibit great variety, we might say that the structure is poorly determined. It might be that there is actually this much conformational variation in this protein model in solution, or that the NMR data did not produce enough constraints to define the structure better.
Prefs: General
Check the box labeled Scale B-factor colors so min = dark blue and max = red. Click OK. This
provides for the maximum contrast in models colored by B-factor.
< shift> Color: B-Factor (hold down shift while executing the menu command—be sure to press shift first)
This command colors in all layers (because of the shift key) according to the numbers in the right-hand or "B-factor" column. In an NMR ensemble, the colors in this column are not B-factors. Instead, the right-hand entry in each atom row contains the deviation, in angstroms, of that atom from the average position of the same atom in the entire ensemble. Atoms colored blue are near the ensemble-average position. Atoms colored red are quite far from the average position of that atom in the ensemble.
Blink continuously through the ensemble (hold down ctrl and tab) and watch a region that is red or orange. You will see that the region does not have the same color in each layer. In some layers, that region is near the average position of the ensemble; in others, the region is quite far away from the average position.
Color: Layer
DeepView assigns a different color to each layer, making it easy to
see the similarities and differences between the models. Rotate the
model to get familiar with it. Interleukin 4 is an unusual four-helix
bundle with two long loops that connect consecutive helices at
opposite ends.
In the helical areas, the 22 models are very
similar. In the loops, there are more differences, while in the chain
termini, the models vary dramatically. This means that very different
conformations can fit the NMR-derived constraints for the termini,
while only similar conformations fit the constraints in the helical
regions.
We might say that the structure is well determined
in the helical regions, poorly determined at the termini. Or we might
say that the termini are very flexible, moving rapidly from one
conformation to another. As a result, many atoms show weak or no
spectral signs of nearness to other atoms, and NMR spectra provide
fewer constraints than needed to define a single conformation.
Roughly speaking, the termini are analogous to "hot" regions in a
crystallographic model, and the helices are analogous to "cold"
regions.
Now load the file 1BBN. DeepView will
automatically place this new model in the same orientation as the
1BCN models. Use the Layers Info window to display 1BBN only. Remove H and side
chains, and reduce the mainchain to CA only.
If you have not already done so,
Prefs: General...
Check
Scale B-factor colors so min = dark blue and max = red.
Color: B-Factor
As with ensemble files, PDB files of averaged NMR models contain no
B-factors. For a given atom, the number in the B-factor column of an averaged model gives
the average (actually, RMS) distance from that atom in averaged model
to that same atom in all the other models. Thus this display shows
you, on the averaged model, how well or how poorly all the ensemble
models agree.
If you want to base an interpretion of the action
of interleukin 4 upon the precise positions of atoms in this model,
you can do so with more confidence if the region of interest to you
is in the blue regions of this model. But if you know that the chain
termini are involved in an interaction between interleukin 4 and
another molecule, this model would give you very little information
about that interaction.
Now add side chains to the display of 1BBN. Your
previous actions have colored the side chains by the same criteria as
the main chain. Notice that, in general, side chains vary more among
the ensemble models (remember that you are looking at a property of
the ensemble displayed on the averaged model), and that side chains
in the interior vary less than those on the surface. These
observations agree with what you saw in crystal structures: interior
residues appear more ordered than do surface residues.
Finally, make sure 1BBN is the active layer, and
examine the PDB file by clicking on the document icon just above the
graphics display. Read lines 50-57 of the file to learn the number
and types of constraints from which this model was derived. The
technical
note for this section tells more about
these indicators of model quality.
9c. Judging the Quality of Homology Models
First, read this Technical
Note: How Structures Are "Determined" By Homology Modeling.
For a good brief discussion of the meaning of such terms as correctness, accuracy, and error in homology models, click HERE. The linked page is a chapter in an old but still useful broad introduction to protein models.
If you do not have any experience handling and comparing
multiple models in DeepView, I recommend that you carry out
Section 11, Comparing
Proteins before trying to examine a homology
model, which will usually come to you as a project file containing the model and the template(s) with which it was made.
Now you will use DeepView tools to search a homology model for errors and structural features that do not make sense. But first, to help us with our evaluation, let's meet the molecule we will explore.
Introducing: Peropsin
An important tool in evaluating a homology model of a protein is other knowledge about the protein. This knowledge includes the protein's cellular environment, its function, and any other evidence (spectroscopy, kinetics) about it. This knowledge also includes the same kind of evidence about the template(s). After all, if the target is similar in sequence to the template(s), then target and template probably share other properties. If the model is not compatible with other knowledge about it and its templates, then something is wrong. To give us realistic tools for our evaluation of this model, here is some background information about the target.
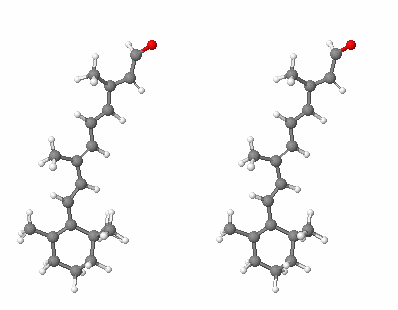
The target of this modeling project, human peropsin (gene name OPSX, UniProt entry O14718), is a protein that is expressed in the rod and cone cells of the human retina. It shares sequence homology with rhodopsin and other human visual pigments (opsins), but apparently does not serve as a light sensor. Its function is not established, but suggested functions include sensing cellular levels of retinal (the light-absorbing pigment found in the visual opsins, pictured below), transport of retinal, or isomerization of retinal during the visual cycle, in which retinal undergoes cis-trans isomerization. (The three-dimensional structure of peropsin is not known, of course; otherwise, we would not need a homology model.)
Retinal, ball and stick model (divergent stereo)
In rhodopsin, the aldehyde group (top) is covalently
bonded to a lysine side chain by an imine link.
I obtained a model of peropsin by submitting a modeling project to SwissModel, specifying as template the ExPDB file 1F88A. This file contains chain A of PDB file 1F88, an X-ray model of bovine rhodopsin, which is the only visual pigment of known structure. Bovine rhodopsin, like the other opsins, lives and functions in the lipid membranes of rod and cone cells. The Protein Data Bank model 1F88 was determined by X-ray diffraction in the presence of detergents, which are often needed to solubilize a membrane protein. Rhodopsin contains a covalently bound molecule of retinal (omitted from the template file). An interesting question to ask about the homology model of peropsin is whether it also appears to contain a binding site for retinal.
For a summary of the latest information about peropsin, click HERE.
Click HERE to download the file that I obtained from SwissModel. Model files like this one come from SwissModel as DeepView project files, so they are ready for exploration in DeepView. Other graphics programs might not be able to reveal all aspects of the file.
Open the downloaded file in DeepView. It consists of two layers, the first named TARGET, and the second named 1f88A, which is the ExPDB template used in modeling.
Explore: Correspondence Between Model and Template
Hold down ctrl and press tab repeatedly) to show the models alternately (called blinking). With TARGET layer showing, turn off display of backbone and side chains, leaving only the colored ribbon (Suggestion: use Prefs: Ribbons to set ribbon display to one strand.) Blink to the template (1f88A) layer, turn off backbone and side chains, and then turn on ribbons. Now blink repeated to compare the two ribbon models. This allows you to see clearly how the backbones of target and template are aligned. Note that the green regions of the target appear almost identical to the template, while the red regions do not correspond to the template. These regions were produced by loop building (a form of guesswork based on loops of similar sequence and end-to-end dimensions in other proteins) or by pure guesswork.
The default ribbon color of a model received from SwissModel is Color: B-Factor, which readily reveals the guesswork regions of a homology model. If guesswork regions correspond to model regions of interest to you in your research on this protein, then you cannot rely on the model without additional evidence that these regions are reasonable.
Explore: Impossible and Improbable Structures in the Model
Blink to make TARGET the active layer. Turn off ribbons, and turn on backbones and side chains.
Select: Residues Making Clashes
Look at the Layers Info window (Prefs: Layers Info) to see if any residues are selected (last column in window). In this model, none are selected, so no residues are in positions that are impossible due to steric hindrance.
(N.B. to GR: would be instructive to build in some clashes and have user fix them.)
Wind: Ramachandran Plot
Select: All
The Ramachandran plot show quite a few residues that are in disallowed conformations (outliers). If you display the template and Select: All, you will see that the template also has some Rama outliers. You cannot expect the target to be better in this regard than the template, but any model is particularly suspect in areas of Rama outliers that are not outliers in the template. Leave the Rama plot available as you carry out the next steps on the TARGET layer.
Color: act on Backbone
Color: Protein Problems
A message tells you the color scheme. All residues are gray except for suspicious ones. Backbone is yellow for bad backbone conformations in non-proline residues, and red for bad proline backbone conformations ("bad" means Rama bad: outliers). Side chains are orange for buried residues whose side chains lack expected hydrogen bonds. Polar side chains in protein interiors are usually joined by hydrogen bonds to other buried polar groups (side chains or backbones). Protein folding replaces most of the lost protein-water hydrogen bonds with internal protein-to-protein hydrogen bonds (usually,  Hfolding ~ 0 kJ/mol). The presence of buried side chains lacking hydrogen bonding neighbors suggests that the residues are placed improperly in the model, perhaps due to faulty sequence alignment with the template.
Hfolding ~ 0 kJ/mol). The presence of buried side chains lacking hydrogen bonding neighbors suggests that the residues are placed improperly in the model, perhaps due to faulty sequence alignment with the template.
To see the residues with problem backbons on the Rama plot,
Select: Residues with same Color as...
Click on any yellow atom in the model (in stereo, click on the left-hand image). Yellow symbols appear on the Rama plot to show the phi and psi angles of the problem residues.
Select: Residues with same Color as... and then click on any red atom in the model (in stereo, click on the left-hand image). Red symbols appear on the Rama plot to show the phi and psi angles of the problem proline residues.
Color: act on Sidechain
Color: Protein Problems
Select: Residues with same Color as...
Click on any orange side chain. Only one residue, ASP213, is selected. Press return to remove all other residues from the display.
Select: Neighbors of Selected Residues
On the Select dialog, click the button for "Select..." and type in 4.5. This adds to the current selection all residues that have at least one atom within 4.5 angstroms of any atom in ASP213.
<return>
Now selected and on display are ASP213 and its nearest neighbors.
Color (Sidechain): Type
You can now see that the red (anionic) aspartate side chain is buried among nonpolar residues, an unlikely situation. If you recolor by protein problems, you will also see that this residues lies in a stretch of Rama outliers.
Coloring sidechains by type makes it easy to see where different side-chain types are located. Base your judgment of side-chain locations on this generality about relative percentages of polar and non-polar residues in protein environments:
Surfaces of water-soluble proteins are predominantly polar; interiors of all proteins are predominately non-polar; membrane-buried surfaces of proteins are the most non-polar of all.
Select: Extend to Other Layers
Blink to the template layer and press return. Now selected and on display are residues of the template that align with the current selection of residues in the target. Notice that fewer residues are on display. Why is that? To find out, add ribbons to both layers and blink repeatedly. You will see that the template is missing some residues in this area. Also note that the target ribbon is red, indicating that this area of the model was built without help from the template.
Now return to the target layer, remove ribbons, display all backbone and side chains, color backbone CPK, and color side chains by type. Also, simplify the image by Display: Show Backbone as Carbon Alpha Trace. With this color scheme, the backbone is white and only shows alpha carbons, making it easy to focus your attention on side chains. "Acidic" side chains (anions) are red, "basic" (cationic) are blue, polar but uncharged are yellow, and nonpolar are gray.
You can see that this protein is largely alpha-helical, and surprisingly, many nonpolar residues lie on the outside (molecular surface) of the helices, especially in the middle of the helices. Is this a problem? Normally, nonpolar residues are buried in protein interiors. But "normally" in this case means water-soluble proteins. Prior knowledge about this protein and its template is useful now. The template, bovine rhodopsin, is a membrane protein, with seven membrane-spanning alpha helices, and a number of turns that project into the aqueous medium on both sides of the membrane. We should assume that this is also true of the target. So the central areas of the helices would be expected to be lipid-soluble, not water-soluble. In fact, the presence of charged residues exposed at membrane-buried surfaces would signal unrealistic areas of the model. Except for the ends of helices, the surface of this model is pretty uniformly nonpolar. The model seems reasonable in this respect. At the ends of the helices, many polar and charged residues face outward, as expected if these regions are exposed to water.
Explore the model, looking for residues that appear in unlikely places. One is ASP47. It is clearly buried, but with no polar neighbors. It was not picked up by Color: Protein Problems, which shows that computer search algorithms cannot always be trusted. If you select and display its neighbors, and then Color (Sidechains): B-Factor, you will see that DeepView applies red color to two residues near ASP47. This means that these side chains have very different properties from the aligned residues in the template. Coloring backbone by "B-Factor" (SwissModel homology models only!) shows how well target fits template. Coloring side chains by "B-factor" reveals residues of the target that align with template residues that are quite different (say, polar versus non-polar, or large versus small).
Another buried residue looks unlikely, at first glance. LYS260 is clearly buried and has no H-bonding neighbors. Compare with the template. You will find a lysine residue (LYS296) perfectly aligned with target LYS260. Here again, prior knowledge can help us to understand. In bovine rhodopsin, LYS296 is the covalent carrier of the retinal prosthetic group. LYS260 of our target appears positioned in precisely the right place to perform the same function. This raises the possibility (which we would need chemical evidence to prove) that peropsin also binds retinal, and in the same manner as does rhodopsin.
Notice how important our prior knowledge is in evaluating this model. If we thought we were looking at a water-soluble protein, and had no idea of the possibility of a lysine side chain covalently bonded to a buried prosthetic group, then a number of perfectly plausible aspects of this model would appear to be errors.
Display the full target model, without ribbons. Open the Alignment window (Wind: Alignment). This window shows exactly how the target and template sequences were aligned before the target was threaded onto the template. Let's take a look at how successful the treading was.
Explore: Other Indicators of Problem Areas in a Model
Color: Threading Energy
The model is now multicolored, with colors near the blue end of the visible spectrum signifying low threading energy, and colors near the red end signifying high threading energy. In the Alignment window, click on the small triangle next to the question mark. The window explands to display a graph of threading energy versus residue number.
Loosely speaking, threading energy tells how "happy" a residue is with its immediate environment. Low energy (blue) is happy, high energy (red) is unhappy. This form of happiness is based on how frequently, in all proteins of know structure, a particular residue is found in a particular environment. It is a good idea to explore model regions of high threading energy to see if there are obviousl problems. Blink to the template, and color it by threading energy. If you activate the graphics window, you can blink to compare the threading-energy graphs of target and template. You will see that the crystallographic model of rhodopsin has a threading energy profile that is very similar to that of the target. So the modeling has not introduced areas where target threading energy is quite different from that of the template, a good sign.
For more information about threading energy, look at the DeepView User Guide (Menus, Color) and the DeepView User Manual.
Finally, let's look at a measure of structural stability throughout the model.
Color: Force-Field Energy
On the dialog that appears uncheck everything except bond and angles. Click OK.
This time the model is mostly blue (low energy, good), with one small red area (high energy, suspicious). The shaky ares is that same low-confidence region revealed by other criteria.
Force-field energy is a real attempt to measure the relative potential energy of each residue in the protein. Low energy means that the residue appears to be energetically stable with regard to the interactions chosen in the Color: Force-Field Energy dialog. Force-field energy can in theory reveal areas of configurational distortion (bad bond lengths and bond angles), conformational distortion (bad torsion angles), or either steric or electrostatic repulsion. Choose the kinds of problems you are seeking by your choices in the Color: Force-Field Energy dialog.
For more information about force-field energy, look at the DeepView User Guide (Menus, Color) and the DeepView User Manual.
Review
Your most important quality-control action is to realize that all "structures"—whether from X-ray crystallography, NMR, homology modeling, or other forms of modeling— are models, not observed structures. Protein molecules are not directly observable. Protein models can harbor many kinds of errors, from minor and possibly inconsequential (subtle distortions of bond lengths, bond angles, or conformational angles) to massive (complete misalignment of the sequence with the evidence [electron density, NMR constraints, or modeling templates]). Practically always, when a model and biochemical data disagree, THE MODEL IS WRONG.
Here are useful command for judging model quality:
- Color: B-Factor is useful for all types of models, but is does not mean the same thing for each type of model. The command colors according to the relative magnitude of the numbers in the far right-hand column of the PDB coordinate file, which was historically referred to as the B-factor column (because, for years, the only PDB models were from crystallography).
NMR and homology modelers use the column for other quality-related parameters, as follows:
- Crystallographers put B-factors (of all things!) in the B-factor column. So for X-ray models, the Color: B-Factor command actually colors by crystallographic B-factor, reflecting the quality of the final electron-density map at each residue, and in a very high-quality model, perhaps reflecting the relative mobility or flexibility of each residue.
- NMR spectroscopists use the right-hand column for a number showing how much the atom positions of a residue vary across the ensemble of models in the PDB file. The number is called the RMS variation for that atom.
- In a homology model from SwissModel, the right-hand PDB file column contains numbers reflecting the entent to which the model is based on the template(s). Blue implies that the target is essentially identical to the template at that residue. Red implies that a residue was not modeled from the template(s), but was made by loop building or other guesswork. Other servers provide similar information
- Many other model-quality tools in DeepView are useful for finding problems with all kinds of models.
- Color: Protein Problems reveals bad backbone conformation and buried polar side chains lacking expected hydrogen bonds.
- Color: Force-Field Energy reveals distortions of configuration (bond lengths and angles) or conformation (torsion angles), as well as unfavorable electrostatic interactions between groups.
- Color: Threading Energy reveals residues that are "unhappy" (not usually found) in their current protein environment.
- Color (Sidechains): Type allows you to see locations of nonpolar, uncharged polar, cationic, and anionic side chains.
- Wind: Ramachandran Plot provides a Ramachandran plot, revealing residues with unlikely backbone conformations.
- Find specific types of unlikely residues with these Select options: Residues Making Clashes, Residues Making Clashes with Backbone, and Sidechains Lacking Proper H-Bond.
- Tools exclusively for X-ray
Models
- The most important quality assessment tool for X-ray models is the electron-density map. Areas of importance to the model user, such as active sites, should have strong, sharp, electron density for all atoms, and the residues should be well fitted to the map.
- Tools exclusively for NMR
Models
- Remember that all models in the ensemble fit the NMR data, so variation among models represents uncertainty in atom positions. Finding a side chain in 10 entirely different positions simply means that we don't know the position of that side chain. You could never guess this from looking at a single model.
- Use Fit: Set Layer Standard Deviation Into B-Factors to color each residue by amount of spatial variation in the ensemble.
- Look at the entire ensemble of models in an NMR file (by blinking). Be suspicious of model regions that vary greatly from one model to the next.
Take time to
PLAY
with the tools introduced in this section.
To gain more expertise in homology modeling, see Gerard Kleywegt's excellent tutorial, Model Validation.
To learn more about homology models, make one! Take the Homology Modeling Tutorial (See ADVANCED TUTORIALS in the Contents frame.) Or learn how to obtain a homology model from an automated server in the tutorial Bioinformatics for Beginners.
Next Section: 10. Working
With Oligomeric Proteins
To The Molecular Level